 |
|
||||||||||
|
|
 |
|
|||||||||
|
|
|
 |
|
||||||||
|
|
|
||||||||||
|
|
|
||||||||||
|
|
|
||||||||||
|
|
|
||||||||||
|
|
|
||||||||||
|
2006, Vol 1 No 1, Article 5
Bovine Spongiform Encephalopathy
and its Relation With Creutzfeld Jakob Disease
Syed Anjum Andrabi INTRODUCTION
Bovine spongiform encephalopathy (BSE) is
a fatal brain disease of cattle. It has long been stated to have been
first observed on a farm in Kent in April 1985, and to have been
identified by Central Veterinary Laboratory (CVL), UK in November 1986
(Wells et al, 1987.) Bovine spongiform encephalopathy (BSE), widely
known as "mad cow disease," is a chronic, degenerative disease affecting
the central nervous system of cattle. World wide there have been more
than 200,000 cases since the disease was first diagnosed in 1986 in
Great Britain. BSE has had a substantial impact on the livestock
industry in the United Kingdom. The disease has also been confirmed in
native-born cattle in Belgium, Denmark, France, Germany, Ireland,
Luxembourg, Liechtenstein, the Netherlands, Northern Ireland, Portugal,
Spain and Switzerland. However, over 95% of all BSE cases have occurred
in the United Kingdom. BSE is not known to exist in the United States.
BSE belongs to the family of diseases known as the transmissible
spongiform encephalopathies (TSE's). These diseases are caused by a
transmissible agent which is yet to be fully characterized.
(b) a progressive debilitating
neurological illness which is always fatal; AETIOLOGY
The causative agent of BSE as well as
other TSE's is yet to be fully characterized. Three main theories on the
nature of the agent have been proposed
Characteristics of the BSE agent. It Prions are responsible for transmissible and inherited disorders of protein conformation. They can also cause sporadic disease, in which neither transmission between individuals nor inheritance is evident. Moreover, there are hints that the prions causing the diseases explored thus far may not be the only ones. Prions made of rather different proteins may contribute to other neuro-degenerative diseases that are quite prevalent in humans. They might even participate in illnesses that attack muscles. The known prion diseases, all fatal, are sometimes referred to as spongiform encephalopathies. They are so named because they frequently cause the brain to become riddled with holes. These illnesses, which can brew for years (or even for decades in humans) are widespread in animals. The most common form is scrapie, found in sheep and goats. Afflicted animals lose coordination and eventually become so incapacitated that they cannot stand. They also become irritable and, in some cases, develop an intense itch that leads them to scrape off their wool or hair (hence the name scrapie). The other prion diseases of animals go by such names as transmissible mink encephalopathy, chronic wasting disease of mule deer and elk, feline spongiform encephalopathy and bovine spongiform encephalopathy. Gerald A. H. Wells and John W. Wilesmith of the Central Veterinary Laboratory in Weybridge, England, identified the condition in 1986, after it began striking cows in Great Britain, causing them to became uncoordinated and unusually apprehensive. The human prion diseases are more obscure. Kuru has been seen only among the Fore Highlanders of Papua New Guinea. They call it the "laughing death." Vincent Zigas of the Australian Public Health Service and D. Carleton Gajdusek of the U.S. National Institutes of Health described it in 1957, noting that many highlanders became afflicted with a strange, fatal disease marked by loss of coordination (ataxia) and often later by dementia. The affected individuals probably acquired Kuru through ritual cannibalism: the Fore tribe reportedly honored the dead by eating their brains. The practice has since stopped, and Kuru has virtually disappeared.
Creutzfeldt-Jakob disease, in contrast,
occurs worldwide and usually becomes evident as dementia. Most of the
time it appears sporadically, striking one person in a million,
typically around age 60. About 10 to 15 percent of cases are inherited,
and a small number are, sadly, iatrogenic-spread inadvertently by the
attempt to treat some other medical problem. Iatrogenic
Creutzfeldt-Jakob disease has apparently been transmitted by corneal
transplantation, implantation of dura mater or electrodes in the brain,
use of contaminated surgical instruments, and injection of growth
hormone derived from human pituitaries (before recombinant growth
hormone became available). The two remaining human disorders are
Gerstmann-Straussler-Scheinker disease (which is manifest as ataxia and
other signs of damage to the cerebellum) and fatal familial insomnia (in
which dementia follows difficulty sleeping). Both these conditions are
usually inherited and typically appear in midlife. Fatal familial
insomnia was discovered only recently, by Elio Lugaresi and Rossella
Medori of the University of Bologna and Pierluigi Gambetti of Case
Western Reserve University. A BRIEF HISTORY OF MAD COW DISEASE
Scrapie, a disease of sheep was investigated more as an oddity and for the interest that it caused specific groups of scientists, particularly in the UK, USA and to a small degree in Germany. Scrapie was thought of as a disease of sheep that did not infect humans, although its tissues were known to contain infection. When BSE arrived, it was immediately thought, because there were no other natural TSEs, to be derived from scrapie and for this to have been fed to cattle in the meal that they ate (to increase their milk yield). By the direction of the UK Ministry of Agriculture, Fisheries and Food (MAFF), a change had taken place in the way that this was made in the early 1980s and this, taking place at a similar time to the original infection of the cattle, was felt to be the answer. A small farm in Surrey reported more than one cow developing a strange neurological disease. The cattle were killed, the brains removed, and the animals destroyed. When it was found that the cattle had a disease never reported before the farmer wanted to publish the data but was reportedly told not to by MAFF. When it is calculated, it seems that approximately 100 cattle had developed BSE symptoms before 1987 and many more would have been infected. It is now suggested that MAFF had been shown cattle with this disease before, and may have known about it in 1983, but did nothing. 1987; the publication of disease and Southwood: Wells et al published the data showing that a cow had developed a spongiform encephalopathy. Little extra data was given. It was clear, however, that MAFF realized that this was no simple disease in that a committee was set up by them to advise on what action should be taken to avoid any risk to humans and cattle. By this time, it was clear that the disease was appearing all over the country. Possibly it spread from the West Country to the other parts but, because of the speed of the spread this was not clear.
Southwood, in the statement that was published stated that there would be minimal risk to humans as all infected cattle would be slaughtered. By not eating the animals with clinical illness there would be no problem and, as the disease was simply scrapie, and scrapie did not spread to humans, we should not expect BSE to spread to any other animals. Humans could continue to eat bovine brain and not worry about the consequences. The answer to BSE was to prevent all bovine material from entering the food that was fed to cattle. This was brought into action in July 1988. The feed manufacturers were warned that this was going to happen several months in advance. The reporting of cases of BSE to MAFF was made obligatory and half the value of a non-sick animal was given in compensation.;
The scientific community was surprised by the relative inaction recommended by Southwood. The committee that was set up by Southwood, known as the Spongiform Encephalopathy Advisory Committee, immediately recommended that specific offals (brain, spleen, thymus, tonsil, gut) should be discarded and that all clinically ill cattle should be destroyed by incineration or burial. By this time BSE had been transmitted to mice in the laboratory and apparently to various zoo animals through the eating of the same feed. Compensation for farmers reporting cases of BSE was only half the value of the cow.
1990; the year of the media hype: The CJD surveillance unit was set up in Edinburgh, UK to find out if BSE was giving rise to extra cases of CJD. The British Parliamentary Agriculture Committee follows a media scare on the risks from BSE. John Gummer, at the time the UK Minister for Agriculture, tries to give his daughter a beef burger in front of the cameras outside parliament (she refused). By this time the numbers of cases were reaching 300 per week. Compensation was stepped up to the full value of the cow and numbers continued to rise. The German Government decided that it would not accept British beef as food in their country because of the risk that it potentially had to their population. Gummer was furious and demanded that less strict laws be taken through the EC Agriculture Committee. The amount of compensation payable to farmers for a case of BSE was increased. Lacey demanded that all infected herds should be slaughtered and that restocking should take place from abroad. Roger Eddy made it clear that he may have seen cases of BSE before the epidemic and suggested that scrapie may not have been its source at all. Gummer made it absolutely clear to the British National Consumer Council that beef was safe and said that there was no risk whatever. A domestic cat develops what was later confirmed as BSE. An American had inoculated scrapie into a cow and it developed a SE, but under the microscope it was not the same as BSE. Various schools ban beef in meals. The centre for agricultural research in Reading demanded that MAFF let professional independent researchers carry out the research into BSE as the results MAFF was releasing led to hysteria. Kiethley News shows that the number of BSE cases was building up so fast that the various parts of the animal could not be incinerated and had to be buried on a local tip. Beef consumption in the UK dropped to the lowest level since 1962. It becomes clear that many of the cases of CJD were never reported. 65% of doctors 'changed their habit of eating beef' due to BSE. All offal banned from export to the EC. A marmoset monkey inoculated with BSE dies.
UK experts were sent to convince them that BSE was not a risk. Harash Narang was reportedly told to stop carrying on his research into BSE and its risk to humans. A case appeared in a cow that was born after the feed ban and they were sure could not have been fed infective material. Fears arose that BSE would also infect the rest of Europe because we had exported infected animals there. The UK would just be ahead of the rest. In the past the knackers would pay 30 for a cow but after 1990 they may actually charge 40 to take it away. The PHLS refused to allow Narang to continue his research. Health and Safety executives bring in directives on how to handle BSE infected carcasses as they might be a risk to the people involved. Watkins the reporter from Today showed that people that had received growth hormone inoculations were still acting as blood donors. It appears that some genetics of a human makes them more likely to develop CJD and have a shorter incubation period. A statutory order from MAFF prevented any use of the specified offals; for a while they had been used for the feed of other animals and as fertiliser. The 'mad calf' syndrome; a calf born to a cow with BSE develops the disease.
A cheetah and the puma died of a TSE now thought to be BSE in the food that they had eaten. It was not clear, however how this could have been through eating brain, as they were never fed this. Fatal familial insomnia is found to be a SE and due to a genetic change. How now mad cow? An editorial in the British Medical Journal saying that we simply did not have enough knowledge to pronounce BSE as safe.
The number of cases was still rising with approximately 800 cases reported in each week. The vets were now being told that many of the cases that they accepted were not actually infected when the animal's brains were looked at under the microscope for evidence of disease; little evidence was ever presented for this and the rate for negatives seemed to remain at approximately 15%. Changes were made in the way that cattle could be sold. The vets that had been at the auctions were decreased in hours and a computer system was organised so that the ear tags on a cow could be used to find out if it was from an infected herd or not. Dealler publishes the data showing that, even using underestimation methods, that the risk to humans was unacceptably high for medical ethics to accept. Farmers were often no longer being asked at the auction if their cattle were from an infected herd and they were receiving better prices from the buyers as a result. Two dairy farmers with BSE in their herds, Mark Duncan and Peter Warhurst, were found to have died of CJD. MAFF claim that there was no infectivity in any tissue outside the central nervous system. A group of chemicals was found that prevented the growth of the infective agent of scrapie in the test tube. The mice without the prion protein gene were grown and found not to be open to infection with scrapie.
Victoria Rimmer, the 16 year old from
North Wales was claimed to be dying of CJD and for this to be due to
having eaten BSE-infected cattle. Cattle meat was being exported for
sale in Europe without evidence that it did not come from a BSE-free
herd. Claims were made that pressure was being put on the vets to sign
certificates without evidence. The start of the Spongiform Encephalopathy Research Campaign: The Germans were unimpressed by 'safe beef pledge' from UK. The EC now made it essential that any meat on the bone being exported could only come from herds unaffected with BSE in the previous 6 years. Gillian Shephard had thought of this as a success and came back and told the newspapers. They quickly realised it was a defeat. A farmer suggests that organophosphorus insecticides may be important in the cause of BSE. CJD reported as being in a similar prevalence in many European countries. It is admitted that, of 156 cases of CJD since 1990, 22 are believed to have given blood at some stage. CJD was found in a butcher from Whitby. Waldergrave takes over as Minister at MAFF. It was shown that abattoirs were attempting to export beef that was from infected herds and that the computer, supposedly carrying the information about all the cattle, was not permitted to give that information out for data control reasons.
It became clear that 1.8 million infected cattle would be eaten from UK farms by the year 2001 and that most of these had already been eaten. Underreporting of cases in 1992 and 1993 was shown to reach 60%. A further farmer died of CJD and a second was dying of what seemed to be the disease. Both were from BSE affected farms. Two teenagers (including Stephen Churchill) developed CJD. Only 4 teenagers had been reported with CJD at any time throughout the world. It now became clear that the feed ban that took place in 1988 was too late. In fact, around 90% of the dairy cattle in the UK turned out being in an infected herd and, due to the apparently limited in-herd rate it seemed that the disease was, by 1988 running out of cattle to involve. If the ban had been in 1987 the number of affected cattle would have been less than half.
March 1996 speeches by Stephen Dorrell (UK Minister for Health) and Hogg (UK Minister for Agriculture Fisheries and Food) admitted that 10 people with a new form of CJD, a variant form, CJD2, had appeared and 8 had died. They must assume that the disease was due to BSE being present in food before November 1989, when the offals ban was introduced. This was followed by a melt down of the credibility of MAFF and the newspapers and TV were determined to make this happen. The European Union quickly banned the export of cattle from the UK and all bovine products and the viewpoint of the Spongiform encephalopathy advisory Committee (SEAC) was that the actions taken so far was probably all that was needed as long as the directions were really carried out. It was soon made clear that these directions were not. The renderers had been permitting tissue from infected cattle to reach further cattle food, the farmers had been permitting cattle with infection to reach human food also. It was admitted on the TV that certain tissues from a cow should not be accepted as being safe unless de-boned in specific places and overseen. The farmers could not take the effect from the EU and demanded that cattle over 30 months not be eaten but slaughtered as if they had BSE when they went to slaughter at the end of their milking lives. This was followed by an uproar in the press and demands from the EU that this was carried out. Hogg backed down and put forward plans concerning major slaughter policies to the EU. They said they were completely inadequate and demanded a greater action. Hogg put up the slaughter from 40,000 to eventually 147,000 but even that was not good enough for some members as they saw no way to get rid of all infected cattle. Mr. Major was determined to complain at the way that the EU had put a ban on the UK beef and refused to allow further EU major action unless this ban was removed after several weeks of the UK preventing EC action, and just before a conference the EC they would permit the decrease in the ban and Major declared that he had won. In fact the EC hardly offered anything and any cut in the ban depended on further demands of the EC at later dates. It became clear after 19th of July SEAC meeting that there had been vertical transmission of BSE between dam and calf. The disease was not likely to leave the UK for many years and, even though MAFF said that 2005 would be good, anyone looking at the figures could see that these were very hopeful. Shortly afterwards it was announced that sheep could easily become infected from BSE and, as we had infected plenty of cattle, there is no reason why sheep could not already be infected. The French quickly put a brain and spinal cord offals ban on all sheep for human consumption and the same may appear in the UK. The prevention of information being released was made clear in a major Nature article in September and this was followed by calls for the release of the information. MAFF agreed, but it has still not happened to any great degree (by Dec 1996). Indications that new variant CJD really was derived from BSE came when Collinge's group in London showed them to be of the same glycoform (i.e. had similar sugar chains attatched). The European Commission had by this time been shown to have deliberately played down BSE and its potential problems all the way through and that there had been a sort of agreement to silence among the Agriculture Commission. The European Parliament found this intolerable and set up a separate committee to look into the matter. This decided that the silence from the EC was intolerable and that many of the official groups were guilty of inaction. Eventually the Governments and the research groups stirred themselves to produce specific research committees and timetables. The dreadful misleading of the population and parliament and the confusion of the farmers by MAFF action, even though they had seen their action as been for the good of farming in the UK, all became much clearer to the population. Even though beef infection had almost been accepted by the UK population, who did not stop to the same degree as those in Europe eating it, it was clear that to get rid of the disease was going to be difficult and may be expected to take many years. By the end of the year it seems that the UK Government have admitted that little beef will be exported for many years and that they will have to carry out the cull of animals that was temporarily dropped by MAFF. Also, the highly expensive actions (mentioned at 3.2 billion pounds by Tony Blair in December 1996) did not take into account the realisation that huge amounts of research were going to have to be done.
The continuous fight by Mr Hogg for the
EC to withdraw its ruling that no UK beef should be exported was
unsuccessful, and visits to the UK by various foreign groups simply made
things worse as they spoke to the farmers who must have seemed simply
misled to the visitors. Hogg would not withdraw his demand, asked
Pattison to go to Brussels to fight for him, and continued to pay the
farmers really quite large amounts of money, realising that after the
election, if the levels of compensation continued it would be in
billions of pounds. The Tory Government was crushed in the election,
which showed the greatest Labour majority ever and a new Prime Minister,
Tony Blair, who was willing to make peace with the European neighbors.
The new Agriculture Minister, Dr. Jack Cunningham, was apparently much
more understanding of the problems than his predecessors and immediately
stated that he did not expect the EC to back down while a risk was
present and the only way out was to remove the risk, initially by
permitting Northern Ireland beef (the area with the fewest cases and
good computer regulation of cattle), and then allowing this to spread to
other areas as they improved. He announced that the costs would be over
3.5 billion pounds and gradually, as the year went on he admitted that
the figure may well turn out to be much greater. The scientific
publication by Moira Bruce that BSE, when inoculated into mice produced
the same disease as nvCJD was the nail in the coffin for the continuing
groups that claimed that the association between the two 'was not
proved' (and therefore should not be assumed). Dolly the sheep appeared
as a sheep made from a single breast cell (named after Dolly Parson, the
singer with the large breasts) and it was realised that the making of
sheep that could not develop BSE or scrapie was at last possible.
Research money arrived for this. The major researchers for MAFF in the
field; Tony Austen and Mike Richards left them and the research money
started to flow into the medical side as the ban on Public Health
involvement in the subject was 'lifted'. The realisation that blood
transfusion could represent a risk hit the press but was overrun by
other subjects. This became a major problem for the Government behind
the scenes, as there appeared to be no way in which UK blood could be
looked on as being totally safe. Eventually the blood products
manufacturers realised that this was indeed true and looked for any way
in which they could keep going and the foreign suppliers moved in. The
Haemophilia society and the Directors of the Haemophilia Centres (the
doctors that proscribe the drugs) made it clear that they did not want
UK factor 8 etc. It was announced that various other tissues would be
included in the list of Specified Bovine Materials: bone, the dorsal
root ganglia, and the lung (the lung part hardly seemed to reach the
press). This was handled well by the new Government and the media ended
up asking why they had banned them at all as the 'risk was so low'. The
announcement that there was to be a public judicial inquiry into BSE and
nvCJD up until 1996 was announced by Jack Cunningham but the scientific
advisors to Lord Justice Phillips are not yet named. They are likely to
be some of the most important people in the inquiry, as they will direct
him towards certain aspects of the epidemic. The aim is to find out why
it all took place, and why such poor action was taken. The inquiry has
to report before the end of 1998. During the year about 12 further cases
of nvCJD appeared, compensation reached approximately 1 billion pounds,
research budgetting was in some quarters put as 'unlimited', and nvCJD
was admitted to be BSE by the UK Government. The developments since 1997 are well known and need not necessarily be included under history.
ORIGIN AND EPIDEMIOLOGY
Various hypothesis have come up to
explain origin of BSE, some of the popular ones include:
Knackers yard greaves involved in
transmission Pro: 1. Cattle could be infected with scrapie by experiment. 2. Epidemiology did not start as a single case and followed by multiple peaks. Anti: 1. BSE was not the same strain of TSE as any scrapie investigated previously. 2. When scrapie inoculated into cows they got a different form of illness. 3. No fall in incubation period was seen. 4. BSE did not have the same range of infectivity as scrapie. 5. No apparent increase in scrapie during the perioddespite large amounts of MBM fed to sheep. 6. Areas where BSE started were not particularly sheep areas (could be explained by movement of sheep carcases to renderers). 7. No clear alteration in infectivity of MBM using solvent extraction methods. 8. No alteration in infectivity expected in MBM due to continuous rather than batch rendering.
9. Epidemiology and clinical nature of
disease suggests that all BSE derived originally from a single point
source i.e. not from many different strains of scrapie. Pro: 1. No fall in incubation period was seen early in the epidemic 2. This would not require the first case to have taken place in 1981 but could have been earlier. 3. No necessity for rendering processes to be involved (no evidence that they they were). 4. Areas where BSE started were not particularly sheep areas (could be explained by movement of sheep carcases to renderers). 5. BSE has a specific range of infectivity (separate from scrapie apparently). 6. No increase in sheep disease was necessarily going to be seen. 7. Outbreak of TME in Stetsonville possibly associated with a TSE in a cow. 8. Reports from veterinary surgeons that they feel they had seen cases of BSE before the epidemic simply as rare conditions in cattle. 9. Work by Collinge and by Bruce indicates that BSE is not similar to scrapie of the strains that they have available. 10. Epidemiology and clinical nature of disease suggests that all BSE derived originally from a single point source. i.e. not from many different strains of scrapie. Anti: 1. The index BSE case would be expected to give rise to BSE cases that were most infective between 3-6 years later and those would be expected to give rise to a second peak 3-6 years after that (each peak broader but 10-100 times higher (Dealler, Donnelly). As such it might be expected to see a specific shape to the epidemic curve. No such shape was seen. This is a quite difficult problem to avoid. 2. Unclear why other countries using similar agricultural methods did not get a similar outbread of disease.
3. If it came from a single animal with
disease then this index case may have been in the early 1970s or 1960s
and could not have been associated with rendering methods. No evidence
for this. Pro: 1. Antibodies were raised for the radio immuno assays for various human pituitary hormones by inoculating the hormones into animals. The animals would have been destroyed at the end of their useful lifespan. The most impure hormones used for inoculation would have been in the 1960s. 2. Body destruction following murder has been suggested to have been by rendering. 3. The spread of human body ashes on land. 4. No requirement for the initial inoculation of cattle to have taken place in 1980. As such it would not be likely to see a series of peaks before the main epidemic rises. 5. Poor evidence that BSE derived from scrapie and no certainty that BSE appeared in UK as a sporadic infection Anti:
1. Work by Collinge and by Bruce showed
BSE not to be similar to CJD of strains that are known but rather to be
similar to nvCJD, which was unknown prior to BSE. (but some aspects of
strain may change when transferred from one species to another) Pro: 1. Similar symptoms to OP toxicity 2. Changes in OPs during epidemic period 3. Same OPs used in specific countries with BSE 4. Warble fly treatment at a specific part of the year and this could explain why BSE is more common in cattle born at specific times. 5. Low rate of BSE in cattle of organic farms that did not use OP. Anti: 1. Epidemiology of BSE does not fit the usage of OP 2. OPs used in specific groups whereas as a toxicity it would be expected not to have a peak at 4-6 yrs. 3. Cattle claimed not to have had any OP exposure develop BSE 4. OPs were in use before BSE appeared.
5. No specific OP changes took place in
1988 when the peak was reached in BSE.
This is a complex hypothesis put forward
by Tom Stockdale and involves the carriage of bacterial toxins to
brain by a glycoprotein receptor corresponding to PrPc. This would
result from the presence in the gut of abnormal proteins (e.g. fish).
This is the complex hypothesis put
forward by Kevin O'Donnell from Edinburgh University. In this he notes
that the PrPsc form of the prion protein builds up partly because it
cannot be broken down by lysosomes. He suggests that sporadic cases and
familial cases may actually not be due to PrP alterations but possibly
because the lysosomes fail to destroy them and they then build up as a
crystalloid structure. The narrow genetic difference among dairy cattle
in the UK might be involved in this.
Horizontal Transmission
Feed Changes
This disease is referenced in old books
as one of a group of ailments known as staggers. About 50 years ago, the
disease was 'lost', possibly because its incidence decreased, and all
references to staggers are now assumed to relate to alkaloidal
poisoning, most commonly by ragwort species (Senecio jacobea and Senecio
vulgaris) in the UK countryside. There is no cure for staggers caused by
alkaloidal poisoning. The symptoms occur when liver damage has reached a
critical and irreversible level. However, the 'lost' disease also
referred to as staggers was considered to be curable, and one cure was
minute doses of ragwort. Giving more ragwort to an animal already
suffering from ragwort poisoning could only hasten its death, but it is
just possible that the specific alkaloids in ragwort could have a
beneficial effect in BSE in very small doses. It is possible that
infected horsemeat was introduced into cattle feed during the 1980s, and
that nvCJD may be transmitted directly from horses to humans, through
vectors such as ticks or faeces. From press reports, at least some nvCJD
victims had links with horses.
Possibly associated with the lysosomal
storage type syndrome of Kevin O Donnell.
The value of the pound at the beginning
of the 1980s was extremely high and the importation of protein for
animal food was felt to be worthwhile. It has been claimed (Independent
1997) that the disease was in fact prevalent in one of the animals from
which the bones were from e.g. gazelles.
The change on bovine diet due to
alterations in the type of source. This may explain why the disease
started when it did in 1980 as it was then that the diet changes took
place. Also there have been many indications that copper deficiency
gives rise to a spongiform encephalopathy although no sign has been
present as to why this is present. The suggestion is that possibly
either copper deficiency sparked the epidemic by making animals
sensitive to a transferred disease or it is more involved in the ongoing
disease process. This is partly described by Axelrad in Medical Hypotheses (March 1998)and by Alan Ebringer in various publications. Pro: The appearance of the brain similar to auto immune disease Chronic process Antibodies to certain bacteria (acinetobacters) higher in cattle with BSE than in cattle without BSE. These bugs produce proteins that cross react antigenically with PrP Minor fall after feed ban. Anti:
Epidemiology does not fit with autoimmune
disease. Acinetobacters are not new and are present in all countries of
the world. Peak at age 4-6 yrs and incubation period relatively steady.
But no reason why this should be so for auto-antibody disease.
Transmissibility of disease by inoculation Fall in the disease when feed
ban introduced. Some farms that do not use certain feeds or OPs do not
also have BSE but they would be expected to have acinetobacters similar
to other farms. No antibodies to PrP found in blood of animals with BSE
or any TSE. Inflammation at the plaque site is present but is not in an
auto immune fashion. No reason why a prion form to a PrP protein should
be involved in any way No experimental data concerning the inoculation
of acinetobacters and the presence of antibodies being associated with
the development of a TSE that is then transmissible. TSE can be
transmitted from one animal to another by inoculation of purified
material that could not contain bacteria. No response to antibiotics:
even in animals given heavy doses from long before symptoms appear.
Animals that cannot produce any antibodies whatever still develop TSE
when inoculated with material. Animals that cannot produce any
antibodies or T-cells still can develop TSEs. Alterations using chemicals
of the immune system affect the incubation period only marginally or the
development of disease (e.g. steroids, anti-cancer drugs). Disease is
modified by amphotericin B, which is not thought to alter the immune
system at all. Infection can be demonstrated in vitro in which no immune
system is present whatever.
This initially seems implausible but the
work done by Done, who indicated that an organism that could be cultured
on agar could be shown to be resistant to all the factors that seemed to
similar to prions. i.e. heat, light, radiation etc. He also showed that
it could be transmitted. His reasearch has been little noticed except by
one group that suggest that the BSE agent was transmitted in the same
way as bovine gut flora in advance of the rumen bacterial flora (Austen,
T) or as a campylobacter-like organism between animals (Roland Heynkes).
The research into it seems to have been refused by MAFF. This sort of
hypothesis can explain the epidemiology of the disease quite well and
the reasons found that cattle born within a few days of each other seem
to share the same risk of BSE on a particular farm. It should be brought
in at this point that the amazing work by Bastian concerning the
possibility that there is an agent for TSEs that are spiroplasma has
been largely ignored in the literature of BSE. Ed Gehrman has put
forward this repeatedly on the net but little has got into the
literature.
These are small, sugar-like compounds
that prevent the breakdown of carbohydrate chains that are on the
surface of proteins. The effect they have mainly is to cause all the
chains to be the same. i.e. when the AGIs are not present, the PrP made
by one cell will carry different carbohydrate chains from the PrP made
by another. When the AGI is added the carbohydrates become the same.
Now, one of the things that is required for the transmission of PrPsc
infection may well be that the carbohydrate chains are identical on the
infecting PrPsc and on the PrPc that is changing into its form. This can
be made certain of by AGIs. Remember, at the beginning of the 1980s
there was a huge increase in the production of foods from potatoes,
which contain large amounts of AGI (Nash et al) and remember that
animals were fed with the potato fractions that were not needed. Also
remember that the sweet potato also contains a lot of them and that the
Kuru affected tribe in New Guinea would have eaten large amounts. (
Further work taking place currently at the University of London.)
The manufacture of methyl bromide and
fluoracetemide from the Rentokil factory at Smarden in Kent gave rise to
chemical release, and groundwater contamination reaching peak in 1963.
Cattle that died as a result of chemical poisoning were sent to knackers
and renderers in the 1960s. The BSE epidemic first appeared 5 miles from
this site. (Abstract from paper: Barry Willis, THIOL-DISULFIDE INTERCHANGE THE KEYTO CONFORMATIONAL CHANGE OF PRION PROTEIN?)
Prion protein is involved in a
group of neuro-degenerative diseases. The conversion of the soluble
cellular prion protein into the pathogenic insoluble isoform is
accompanied by a decrease in a-helical structure and an increase in the
b-pleated sheet content which aggregate into amyloid fibrils. The
conversion of a-helices of prion to the b-pleated sheets is most likely
to result from the reduction of stability of the a-helices relative to
the b-pleated sheet. Such a mechanism of stability reduction exists due
to the presence of the disulfide bond between two a-helical components
of the cellular prion protein and their ability to undergo thiol-disulfide
interchange.
This was particularly of significance in
countries outside the UK and the bovine growth hormone (BGH) was not
licensed for use there. However it is not at all clear that it was not
imported and used illegally and, as a farmer's cattle were valued for
breeding according to the quantity of milk that they produced (BGH
causes milk production) it would be reasonable to consider that it did
take place here. Knowing that HGH caused a spread of CJD from rare human
cases it is not unreasonable to consider that BGH might do so in cattle.
This hypothesis would require the BGH being infected with small amounts
of BSE and also that there was another change in the UK that caused the
epidemic to spread, whereas it did not do so in other countries. BGH use
took place after the second war and rose markedly during the 1970s (but
not in the UK legally). There are now good indications that bovine
pituitary hormones were used for both GH and as a method for embryonic
transfer and implantation in the UK herd. The way in which BSE seemed to
start in the South East of the UK but cases appeared suddenly throughout
the UK is odd and the easy transfer of pituitary hormones around the
country could explain this. The sudden rise in the cases of BSE long
after the use of MBM started was strange and was the fact that the
numbers rose so rapidly whereas if BSE was derived from a single
sporadic BSE case we would have expected a different growth pattern of
disease. The subject is fully discussed in the Phillips Inquiry and the
explanations are fairly convincing as the method of transfer of the
original cases to get the numbers up to a level to cause the epidemic.
Farmers have been fairly clear that they did use these hormones but it
is unlikely that they will make this public.
According to the hypothesis the causer of
the disease is oligonucleoprotein, that is situated in the genome of the
host. In fact, the causer is a nucleosome which is constucted of
oliginucleotide and histones. It differs from nucleosome by the
succession of nucleotides and this very difference is the cause of the
pathology. That infectious oligonucleoprotein integrates into the
construction of the host genome. After being integrated into the host
genome, it makes changes in the construction of the gene and if it is a
unique gene, then the primary construction of the corresponding protein
changes. The gene mutation caused by the integration of the infectious
oligonucleoprotein is a reason for an anomaly constructed enzyme
production. The mutation of the given gene may occur spontaneously too
as well as the mutation may be inherited. The causer of the disease is
not inactivated by DNAse because DNAse ruins DNP/P-protein/ up to
oligonucleoprotein, which is resistant. According to the hypothesis the
causer is the stated above oligonucleoprotein, which is resistant to the
DNAse inactivising influence and vice versa, after the influence of
DNAse the causer separates itself from the genome and may produce
infection.The DNAse cannot ruin the oligonucleoprotein, because the
molecules of the histones protect it. But the causer may be inactivated
by PROTEase, because the PROTEase destroys histones and they don't
protect anymore the oligonucleotide of the destruction. Furthermore ,prions appear
to remain infectious even after being exposed to treatments that destroy
nucleic acids. Besides, the fact that in PH=10 condition the causer is
inactivated, it also agrees whit the hypothesis. As the histones are
alkaline proteins, then in PH=10 condition they are deproteinated/they
lose H+ ion/,they lose the positive charge, resulting the lost of ionic
contact between negative charges of phosphates of nucleotid and positive
charges of the histones. That is histones separate themselves from
oligonucleotides. The hypothesis also gives explanation to the question
how the multiplations of the causer takes place. Antibodies don't
produce to oligonucleoproteins, because last one have small sizes. (This
hypothesis requires further information to justify various interactions
with PrP and to explain the many scientific findings with the disease)
Some reason was needed to work out why
cattle seemed to become infected before 18months of age. Statistically
it seems that they are either infected between 6 months and 18 months
or, to a large degree before or shortly after birth. As there seems to
be little reason why 6-18 months should be the reason at all, it has
been considered that almost all the cases that we see are in fact
infected from their mother. For some reason the proportion of cattle in
any herd that develop BSE rises quickly to around 15% but uncommonly
gets any further. Because of this the epidemic in case numbers that is
seen is not so much due to an increase in cases in herds but an increase
in the number of herds that are affected. The question arises as to why
only 15% should be affected and this is unclear; why not 100%? Surely
cattle would be more likely to be exposed to BSE in their milking life
when they were fed larger quantities of MBM, but it is not these cattle
that seem to develop any disease but rather ones exposed early in their
life. The hypothesis suggests that when a herd is infected, in fact it
is a very large proportion that do. However they would develop symptoms
(if at all) late in their life (perhaps aged 17) i.e. long after
slaughter. As such the proportion that we see developing symptoms are
the offspring of those mothers and that the VT rate is 15%. The
epidemiology for this is not unreasonable but involved Ro factors
dropping long before the feed ban was introduced. Fergusson's work at
Oxford suggests that the Ro did in fact drop in association with the
feed ban although the refusal of MAFF to permit any other group but this
one to work on the data makes this difficult to argue.
This was put forward by Dr. Jane Karlsson
and goes through how it is not really surprising that certain symptoms
are involved in UK animals and perhaps may be particularly susceptible to
BSE in that neurotoxic heavy metals and interactions between them are
repeatedly discussed as being of importance in the bovine diet of the
UK.
This follows the ideas of Arne N Gjorgov,
who published an article 'Prevention and eradication of the BSE: A
solicited response and proposal' Macedonian Vet Rev. 1996;25:97-101. It
follows the fatal neurological condition of pseudopregnant mice
following sterile mating whereas none of the controls that received
prostaglandins developed the condition. The hypothesis is that this kind
of disease may be transferred through a mating system in the mice and
prevented by prostaglandins. It is suggested that prostaglandins may be
a prophylactic factor in BSE.
Another view is that outbreaks of actual
disease occur when an otherwise silent C-DNA resulting from initial
accidentlal retrotransfection of RNA semi-virus is activated into
re-transcription. The question to think is why so many cows and people
do NOT get BSE/CJD once infected and whether this happy state can be
prolonged throughout life. Part of this hypothesis therefore is that
"environmental" affronts whether by atmosphere (insecticides) of food
may activate "disease" from benign latency. Queniborough is the next
village to Syston where Asmer Flower Seeds greenhouses were overcrowded
with hanging insecticide impregnated yellow terrors. How many at
Queniborough worked at Syston? Was there or is there any intensive
horticultural glasshouse nursery near the Doncaster street where the two
new occurrences come from. Did such nurseries if they exist also used
toxic hanging strips at high intensity? This idea of course also relates
to sheep dips etc. The hypothesis explores specific reasons why this
should be so and how the PrP becomes changed as a result. Some of this
relates to the ability to immunise an animal with one scrapie strain
against another. The idea will explain a number of things such as the
rapid spread of BSE in the UK but the low percentage of cattle that
actually die per herd.
By 1994 it had become clear that it was
difficult for the initial scrapie hypothesis to be accepted and that the
'sporadic BSE' hypothesis was more reasonable. However, by 1995 the
findings by Taylor et al that the changes in the rendering processes
could not be clearly seen as the cause of the epidemic and the other
factors in the discussion above made it clear that a further examination
was required. Without the rendering changes being involved, the index
BSE case would have had to have taken place in the early 1970s or late
1960s. Because of the problems concerning the source of BSE from a
single case a further hypothesis is considered as below in conjunction
with Dr. A. Maddocks: "BSE is derived from a random disease in cattle,
it is not adequately destroyed by MBM rendering and it is fed to cattle.
This has taken place for many years but will only be in adequate doses
when UDP feeding techniques (UnDegraded Protein) are used (which were
introduced progressively as of 1980). It is the offspring of these
cattle that develop the symptomatic disease having become vertically
infected." This hypothesis would not require repeated peaks after an
index case, it would not require the animal to have been fed any
infective meal and it would not expect large numbers to become
symptomatic from a single source. Also, it would expect to produce a
drop in Ro early in the epidemic, without large numbers being apparently
infected (although they really were). It also explains the relatively
slow (after correcting for under reporting) fall in BSE cases in cattle
born after the feed ban. The reason that the hypothesis was put forward
was because it answered the problems listed in 'discussion' above. The
only clear problems with the hypothesis are that it cannot explain why
few cases of BSE have been diagnosed in Europe* and that several cattle
(about 20) would have been infected with BSE at the point at which UDP
was introduced (not a single cow). A statistical model has been made of
this hypothesis but not published at this time. *NB sheep exported from
the UK did develop scrapie in New Zealand and Australia and were
genetically the same range as the ones that developed the disease in the
UK. However the no further sheep, including the offspring of the
infected sheep developed the disease. This suggests that an
environmental factor is involved and the possibility must be considered
that a similar factor is also involved in BSE in Europe.
Logic: Rapid drop in cases of BSE born in
the short period before the 1988 feed ban shows infection to be taking
place within first 6 months and mostly within the first month of life.
(not under argument now) Age distribution of cattle with clinical BSE
did not change as the epidemic progressed: therefore they must be
getting infected with a similar dose throughout the epidemic. Extremely
low amounts likely to be found in the MBM anyway of prions. Cattle gut
mucosa will absorb large molecules easily after birth but this effect
disappears within the first few months. It is thought that the effect is
to permit the uptake into the bloodstream of mother's antibodies from
colostrum. The mucosal resistance to this builds up and is thought to be
caused by the change in diet after around 1-4 weeks of life on the farm.
This would be expected to cause the dose size of BSE reaching the blood
of the animal to decrease markedly as the calf gets past the first few
days of life and hence the incubation period would be expected to
increase. Therefore it is not surprising to find a wide range of age
distribution in the cattle with clinical BSE. Cattle developing BSE when
born on farms where no MBM was used but brought from other farms and a
large proportion of the calves were not fed any MBM but were suckled
until relatively old (particularly in beef farms). Cattle born within a
month of each other share the chance of developing BSE to some degree.
This could be perhaps because of feed (feed deliveries were often a
month apart), or because of contamination that was washed away at a
later date. These findings suggest that some factor is not exposed to
all the cattle born at roughly the same time (e.g. common mixed
colostrum) but affect a small proportion. The suggestion is that the
infection is either present in environmental contamination to which the
calves are exposed and is washed away, or to feed contamination that
will only infect cattle if young enough to get past their gut mucosa. As
a result of this a wide age distribution would be expected for clinical
cases and no change in age distribution expected as a result of the feed
ban. CATEGORISATION OF POTENTIAL INFECTIVITY OF DIFFERENT ORGANS IN BSE AFFECTED ANIMALS
Categorising the potential infectivity of
different organs in BSE-infected animals. The assessment of the
infectivity is based in part on scrapie titres, on the finding of high
infectivity in the brain of BSE-affected cattle, on the differential
impact of BSE-infective organs on the infection of mice to intracerebral
inoculation and on the presumed CJD infectivity of human dura mater and
human pituitary gland based on transplant data and the effects of human
growth hormone infection. For practical reasons relating to
slaughterhouse contamination, some tissues are categorised at a higher
level than warranted by their intrinsic infectivity. PATHOGENESIS The "infectious" prion protein is felt to be an abnormally folded version of a protein that is normally found in every animal specie, called PrP. In other words, it is a single protein chain that is just put together in an unusual way. The abnormally folded or knotted version of the prion protein has some unusual and remarkable characteristics. First, it is extremely stable, surviving conditions that would destroy most proteins. Second, it seems to have the ability to cause other normally folded prion proteins to adopt the abnormal folding pattern. Imagine that some abnormally folded prion protein got into your brain. Since it appears to be able to cause the normal protein to become abnormal, i.e. through propagation of an abnormal structure, you can imagine that over time you would start to have an accumulation of the abnormal protein in your brain. How could that abnormally folded protein get into your brain to start with? Well, you could come in contact with the brain of a person or animal that has an accumulation of abnormal protein in their brain. Once it found its way to your brain (?by eating it?), the slow process of accumulation could begin. Otherwise, some PrP in your brain may just spontaneously fold abnormally, i.e. by itself. In fact, there are some human mutations that can cause this abnormal transition to happen with increased frequency, leading to inherited forms of CJD that do not require exposure to an infected person. In the end, there is a lot of evidence to support the notion that the infectious prion agents act by the transmission of an abnormal protein structure, but this is not yet formally proven. Even if you accept this hypothesis, it still falls short of explaining how prion proteins actually cause neurological disease! In other words, why does an accumulation of abnormally folded prion protein cause neurodegeneration. There is no clear answer to this question yet. It does not appear to be due to loss of function of the normally folded protein. Instead, it seems more likely that the abnormal protein is in some way toxic, causing neurons to die or make ineffective and inappropriate connections. The one thing that is certain is that the brain eventually becomes highly disorganized in its activity, and to degenerate physically, which signals the beginning of the end. Evidently, the scrapie protein propagates itself by contacting normal PrP molecules and somehow causing them to unfold and flip from their usual conformation to the scrapie shape. This change initiates a cascade in which newly converted molecules change the shape of other normal PrP molecules, and so on. These events apparently occur on a membrane in the cell interior. The differences between cellular and scrapie forms of PrP must thus be conformational since other possibilities seem unlikely. For instance, it has long been known that the infectious form often has the same amino acid sequence as the normal type. Of course, molecules that start off being identical can later be chemically modified in ways that alter their activity. But intensive investigations by Neil Stahl and Michael A. Baldwin in USA have turned up no differences of this kind.
Fig 2: Scrapie (Sheep) Fig 3: Kuru (Man) Fig 4: CJD (Man)
Electron Micrographs of brains carrying TSE's
CLINICAL SIGNS
The affected animals may display changes
in temperament, such as nervousness
or aggression; abnormal posture;
in-coordination and difficulty in rising; decreased milk production;
or loss of body condition despite
continued appetite. There is no treatment, and affected cattle
die
DIAGNOSIS
There is no test to detect the disease in a live animal. Microscopic examination of brain tissue at necropsy is the primary laboratory method used to confirm a diagnosis of BSE. There are also several techniques used to detect the partially-proteinase resistant form of the prion (PrPres) protein. These techniques are immuno-histochemistry and immuno-blotting.
RELATION BETWEEN BSE AND CJD
CJD is a slow degenerative human disease of the central nervous system with obvious dysfunction, progressive dementia, and vacuolar degeneration of the brain. CJD occurs sporadically worldwide at a rate of 1 case per 1 million people per year. There is a strong epidemiologic and laboratory evidence for a causal association between new variant CJD and BSE. The absence of confirmed cases of new variant CJD in other geographic areas free of BSE supports a causal association. In addition, the interval between the most likely period for the initial exposure of the population to potentially BSE contaminated food (1984-1986) and onset of initial new variant CJD cases (1994-1996) is consistent with known incubation periods for CJD. An experimental study reported in June 1996 showed that three cynomologus macaque monkeys inoculated with brain tissue obtained from cattle with BSE had clinical and neuropathological features strikingly similar to new variant CJD (Nature 1996;381:743-4). A study published in 1996 indicated that a Western blot analysis of infecting prions obtained from 10 new variant CJD patients and BSE-infected animals had similar molecular characteristics that were distinct from prions obtained from patients with other types of CJD (Nature 1996;383:685-90). Most recently, interim results of an ongoing experimental study involving inoculation of a panel of inbred mice with the agents causing BSE and new variant CJD substantially increased the strength of the scientific evidence for a causal association between new variant CJD and BSE (Nature 1997;389:498-501). The incidence of classical CJD in the United States (about 1 case per 1 million population per year) is similar to the incidence found in the rest of the world, which includes Australia and New Zealand--countries that have NOT reported either scrapie or BSE. CJD, which was first diagnosed in the 1920's, occurs with roughly the same frequency in Britain as in other countries at the present time. On March 20, 1996, the UK's Spongiform Encephalopathy Advisory Committee (SEAC) announced the identification of 10 cases of a new variant form of CJD (vCJD). All of the patients developed onset of illness in 1994 or 1995. The following features describe how these 10 cases differed from the sporadic form of CJD: The affected individuals were much younger than the classical CJD patient. Typically, CJD patients are over 63 years old. The average patient age for the onset of variant CJD is 28 (range of 14 to 52) years. The course of the disease in the vCJD averaged 13 months. Classical CJD cases average 6 month duration. In the variant cases, electroencephalographic (EEG) electrical activity in the brain was not typical of sporadic CJD. Although brain pathology was recognizable as CJD, the pattern was different from sporadic CJD, with large aggregates of prion protein plaques. Epidemiological and case studies have not revealed a common risk factor among the cases of vCJD. According to the SEAC, all victims were reported to have eaten beef or beef products in the last 10 years, but none had knowingly eaten brain material. One of the affected individuals had been a vegetarian since 1991. The SEAC concluded that although there was no direct scientific evidence of a link between BSE and vCJD, based on current data and in the absence of any credible alternative, the most likely explanation at that time was that the cases were linked to exposure to BSE before the introduction of control measures, in particular, the specified bovine offal (SBO) ban in 1989. Research reported in later 1996 and 1997 has found evidence to further support a causal association between vCJD and BSE. Two significant studies published in the October 2, 1997 edition of Nature lead the SEAC to conclude that BSE agent is highly likely to be the cause of vCJD. Dr. Moira Bruce and colleagues at the Institute for Animal Health in Edinburgh, Scotland inoculated 3 panels of inbred mice and one panel of crossbred mice with BSE, vCJD and sporadic CJD. Interim results indicate that mice inoculated with BSE showed the same pattern of incubation time, clinical signs and brain lesions as mice inoculated with tissues from patients with vCJD. This provides evidence that BSE and vCJD have the same signature or are the same "strain". In addition, sporadic CJD and known scrapie strains were not similar to vCJD or BSE. Results from another study published by Dr. John Collinge and colleagues of Imperial College School of Medicine, London, UK strongly support Bruce's results. Collinge's paper reports findings of BSE transmission to transgenic mice expressing only human PrP. Another paper by Collinge et. al. in the October 24, 1996 edition of Nature also provides data to support the association between vCJD and BSE. More recently, studies using transgenic animals expressing the bovine PrP have supported the view that BSE infected cattle are responsible for vCJD. These mice not only propagated the BSE infectious agent in the absence of a species barrier, but also were highly susceptible to vCJD and natural sheep scrapie. Furthermore, the transgenic mice inoculated with either vCJD or BSE had indistinguishable disease characteristics.
TREATMENT AND PREVENTION
Since the nature of the pathogenic organism/material is obscure no currently developed treatment can be effective in this disease. Its only when research has thrown enough light on the aetio-pathogenesis of the disease that a rational treatment can be worked out. Till then the major push will be towards control and eradication of the disease. Some measures taken to control disease in many countries are as under: In July 1988, the UK banned the use of ruminant proteins in the preparation of animal feed. The use in the food chain of bovine offals considered to pose a potential risk to humans was also banned in the UK in 1989. The list of banned bovine offals was revised and expanded on several occasions as new information became available. In other countries, including those in Europe, measures taken, the date of implementation and the extent of enforcement vary from country to country. Starting in 1996, bans prevented the sale of food and food products containing beef from the UK to other countries. Other products (e.g. tallow, gelatin) derived from bovine tissues were also prohibited from sale from the UK to other countries. However, in 1999 the EU lifted the ban for meat fulfilling specific requirements; for example, de-boned beef from animals from farms where there have been no cases of BSE and where the animals are less than 30 months of age at slaughter. Cattle are continuously monitored for BSE and BSE is decreasing in the UK. The number of reports of BSE in the UK began to decline in 1992 and has continuously declined year by year since then. New monitoring programmes using newly developed tests for the diagnosis of BSE in dead cattle have been introduced in Switzerland and France, and may be expected throughout the EU.
WHO CONCLUSIONS AND RECOMMENDATIONS TO REDUCE EXPOSURE TO THE BSE AGENT
All countries must prohibit the use of ruminant tissues in ruminant feed and must exclude tissues that are likely to contain the BSE agent from any animal or human food chain. BSE eradication was recommended during a WHO consultation held in December 1999. All countries are encouraged to conduct risk assessments to determine if they are at risk for BSE in sheep and goats. It is advised that any tissue which may come from deer or elk with Chronic Wasting Disease (CWD, a transmissible spongiform disease of North American mule deer and elk) is not used in animal or human food; however, at this time there is no evidence to suggest that CWD in deer and elk can be transmitted to humans. No infectivity has yet been detected in skeletal muscle tissue. Reassurance can be provided by removal of visible nervous and lymphatic tissue from meat (skeletal muscle). Milk and milk products are considered safe. Tallow and gelatin are considered safe if prepared by a manufacturing process which has been shown experimentally to inactivate the transmissible agent. Human and veterinary vaccines prepared from bovine materials may carry the risk of transmission of animal TSE agents. The pharmaceutical industry should ideally avoid the use of bovine materials and materials from other animal species in which TSEs naturally occur. If absolutely necessary, bovine materials should be obtained from countries which have a surveillance system for BSE in place and which report either zero or only sporadic cases of BSE. These precautions apply to the manufacture of cosmetics as well.
|
|
||||||||||
|
|
|||||||||||
|
|
|||||||||||
|
|
|||||||||||
|
|
|||||||||||
|
Copyright © Vet Scan 2005-2006 All Right Reserved with VetScan and
www.kashvet.org |
|
||||||||||
| Home | e-Learning |Resources | Alumni | Bird Flu | Forums | Disclaimer |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
 3-D
Shape of Prion
3-D
Shape of Prion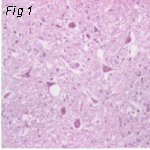
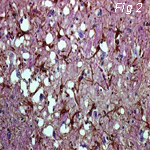
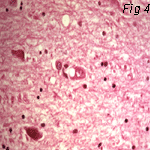
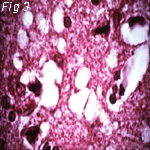 Fig 1: BSE
(Cow)
Fig 1: BSE
(Cow) .
The incubation period ranges from 2 to 8
years. Following the onset of clinical signs, the animal's
condition deteriorates until it dies or is destroyed. This usually
takes from 2 weeks to 6 months.
Most
cases in Great Britain have occurred in dairy cows between 3 and 6
years of age.
.
The incubation period ranges from 2 to 8
years. Following the onset of clinical signs, the animal's
condition deteriorates until it dies or is destroyed. This usually
takes from 2 weeks to 6 months.
Most
cases in Great Britain have occurred in dairy cows between 3 and 6
years of age.